
COVID-19 Vaccine Update
by Bob Kordella, R.Ph., MBA, Chief Clinical Officer
As several COVID-19 vaccine candidates work their way through the clinical trials process, many new terms have begun to work their way into our everyday conversations. Let’s take a few moments to define some of those new terms.
Let’s start with some basics. Immunity is simply protection from an infectious disease. If you are immune to a disease, you can be exposed to it without becoming infected. A vaccine stimulates your immune system to produce antibodies, exactly like it would if you were exposed to the disease. After getting vaccinated, you develop immunity to that disease, without having to get the disease first. This is what makes vaccines such powerful medicine. Unlike most medicines, which treat or cure diseases, vaccines prevent them.
A “clinical trial” is an assessment of the benefits of a new treatment vs. a comparator treatment that occurs in humans, while a “pre-clinical trial” typically occurs in animals. Clinical trials themselves have their own jargon, such as:
- Placebo: A non-active treatment that is used to compare vs. an active treatment under investigation. When a placebo is used a clinical trial is said to be “placebo-controlled.”
- Blinded: When a participant (either the physician and/or the patient) in a clinical trial doesn’t know whether an active treatment or a placebo is being administered/received. “Single blind” usually means that the patient doesn’t know, but the physician does know; “double blind” usually means that neither the physician nor the patient knows.
- Phase I: A Phase I clinical trial is primarily concerned with assessing the safety of a new treatment for use in humans.
- Phase II: A Phase II clinical trial is primarily concerned with establishing a safe dosing range in humans, while continuing to assess safety for use in humans.
- Phase III: A Phase III clinical trial is primarily concerned with establishing that a new treatment does what it is proposed to do, at the established dose, while continuing to assess safety for use in humans.
- Statistical significance: This term implies using sophisticated math tools to objectively determine whether or not a new treatment delivers more clinical effect than a placebo.
Traditional vaccines consist of entire pathogens (e.g. viruses, bacteria) that have been killed or weakened so that they cannot cause disease. Many vaccines in use today fall into this category. However, today new, non-traditional approaches in vaccine development are being used to better target specific components of a pathogen that stimulate the immune system’s response. Vaccines themselves have their own jargon:
- Inactivated vaccine: Also known as a “killed vaccine,” this type of vaccine is produced by first killing a pathogen with chemicals, heat, or radiation. Since the pathogen is dead, it cannot cause disease.
- Live-attenuated vaccine: These vaccines contain a version of the living pathogen that has been weakened in the laboratory, are more common for certain viruses, and typically confer lifelong immunity following only one or two doses. The measles/mumps/rubella (MMR) vaccine is an example.
- Subunit vaccines: Instead of the entire pathogen, subunit vaccines include only the components of a microbe that best stimulate an immune response, such as a particular protein. Although this design can make vaccines safer and easier to produce, it often requires the incorporation of adjuvants (defined below) to elicit a strong protective immune response because the subunits alone are not sufficient to induce adequate long-term immunity.
- Nucleic acid vaccines: This approach introduces genetic material, either DNA or messenger RNA (“mRNA”), into the body that causes the body’s own cells to produce the antigens against which the immune response is sought.
- Viral Vector vaccines: Viral vector vaccines combine many of the positive qualities of nucleic acid vaccines with those of live-attenuated vaccines. This type of vaccine is generally able to produce a stronger immune response than a nucleic acid vaccine alone.
- Adjuvant: A substance that is added during production to increase the body’s immune response to a particular vaccine.
Whew! Lots of jargon, I know, but now that you have a handle on all of it, the following chart that describes the current status of the five COVID-19 vaccine candidates under clinical trial development in the US Government’s “Operation Warp Speed” (OWS) Program will make much more sense. (Six vaccines have been funded by OWS, but one has not yet entered clinical trials.)
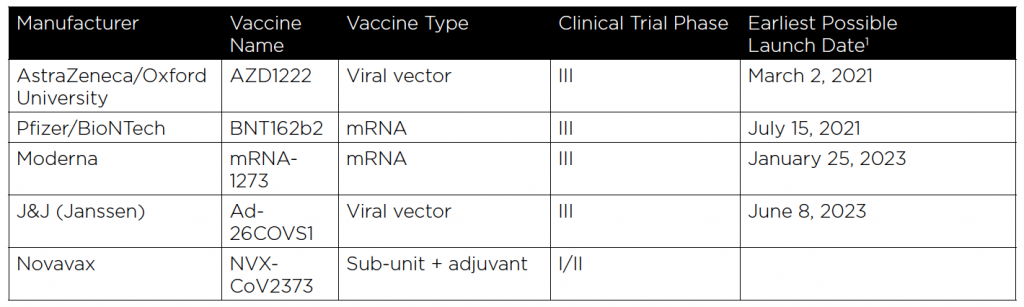
Three more things must occur before victory can be declared in the development of effective COVID-19 vaccines:
- Phase III trials must establish safety and efficacy
- Manufacturers must build or buy large-scale manufacturing capacity
- FDA approval must be granted
The FDA has provided guidance that specifies the objective standards that they will use to grant approval to all vaccine candidates. At a high level the guidance indicates that, “…the FDA would expect that a COVID-19 vaccine would prevent disease or decrease its severity in at least 50% of people who are vaccinated.” (Link to the complete FDA guidance on COVID-19 vaccine approval may be found here.)
The success of multiple vaccines will be important in the event that Phase III trials establish that multiple vaccine doses of a single vaccine will be required to convey immunity, or in the event that combining vaccines of differing types in a series of multiple injections is found to be optimal.

Bob Kordella, R.Ph., MBA
Chief Clinical Officer Vice President
Bob has more than 35 years of diverse experience in the pharmacy industry. Over the course of his career, Bob has led clinical and PBM operations teams in successfully managing more than $4 billion in annual drug spend. This was also while limiting per-member-per-year spending growth to levels that have simultaneously drawn industry acclaim and consistently high levels of member and payer satisfaction.
Hemophilia A –
Is there a Paradigm Shift Ahead?
by Rebecca Lich, PharmD, MBA
and Allison Loi, PharmD Candidate May 2021
Hemophilia is a relatively rare condition, impacting 20,000 – 33,000 Americans. Although hemophilia A and B are the two main subtypes, hemophilia A is about four times more common than hemophilia B, and up to 60% of those affected have the severe form of the disease. These individuals may experience spontaneous (without trauma or injury) bleeding into the soft tissue, joints, or muscle two to five times per month. Therefore, treatment plans include strategies to prevent, and quickly stop bleeds. Often the cost of care is $200-$300K per year, with costs increasing with disease severity, comorbidities, and inhibitor development.
Hemophilia has always been a costly and complex disease state, but with gene therapies, such as Roctavian, around the corner, this disease state has demanded even more attention lately. In fact, many expected Roctavian to become the world’s most expensive drug with a price tag of $2-3 million for a single infusion! However, in a surprise move in August 2020, the FDA is now requesting an additional two years of data before the drug will be approved. There are unanswered questions around the durability of the therapy to know how long it will be effective.
Most individuals who get hurt from a minor injury, such as a cut or scrape, can easily form a clot and stop the bleeding. However, hemophiliacs lack or don’t make enough of an essential protein called clotting factor, which is needed for normal blood clotting. Even small injuries can result in serious health problems for these individuals. Currently, there is no cure for hemophilia. The standard of care for hemophilia A is factor replacement therapy or Hemlibra® through regular infusions and injections, respectively, to replace the missing clotting factor proteins in the body. There are currently many standard and extended half-life recombinant factor VIII products on the market to treat and prevent bleeds. For preventative treatment, most standard factor VIII products are dosed two to four times per week, while longer-acting products are dosed one to two times per week.
New gene therapies attempt to reduce the burden that regular infusions can place on patients and caregivers. Initial research of Roctavian, a gene therapy developed by BioMarin Pharmaceutical Inc., suggests that one high dose of Roctavian may eliminate the need of regular infusions for up to 4 years. This gene therapy works to replace the faulty gene needed to produce factor VIII, which is responsible for hemophilia A.
The cost of managing patients with hemophilia varies with disease severity, frequency of bleeding events, and presence of inhibitors. Hemophilia A treatment is often $200-$300K per year, with inhibitor development pushing treatment costs to over $1 million. About 20% – 30% of people with severe hemophilia A will develop inhibitors (antibodies) against their factor replacement infusions, which increases their risk of bleeding and complications, making it more challenging to treat.
With the proposed pricing of $2-$3 million, Roctavian would be the world’s most expensive one-time therapy. BioMarin Pharmaceutical Inc. argues that the high price tag is justified considering the price in the context of the cumulative costs of managing a patient with hemophilia over his/her lifetime. Preliminary findings from the Institute for Clinical and Economic Review (ICER) suggest that a $2.5M price tag could be cost effective in certain scenarios. . There are 4 other gene therapies in various stages of approval in the pipeline for the treatment of hemophilia A. The expected approval dates for these are 2021 or later. Since the FDA is now requesting additional data before approving Roctavian, there may be impact to the broader pipeline as well. There are inherent challenges in orphan drug research, since there is such a small study population. There are also unanswered questions about long term durability. Cost & care tactics will continue to be essential and include: prior authorization, case management services, inventory management, assay management, site of service, and contracting & network strategies. Gene therapies appear to be great candidates for value-based agreements, but these create their own challenges. Some vendors intend to add these medications to risk-pooling programs, at a PMPM fee to employers.
Hemophilia will continue to be a challenge for plan sponsors and patients due to its medical complexity and cost of care. However, gene therapy could create a paradigm shift in the treatment of hemophilia as we know it.

Rebecca Lich, PharmD, MBA
Senior Vice President
Rebecca is a Doctor of Pharmacy. She is an expert in contract development, pharmacy claim pricing, benefit design, rebate management programs, financial analysis, auditing, and adjudication of pharmacy claims. She has a proven track record in relationship management and people leadership. With more than 10 years Pharmacy Benefit Management experience, including product development, account management, and Sales, Rebecca and her team supported large regional and National health plans with their clinical strategy and PBM operations. This included formulary and plan design, rebate strategy and operations, specialty drug management, clinical programs, and solving complex benefit issues.
Allison Loi, PharmD Candidate May 2021
Pharmaceutical Manufacturer
Lawsuits Impact Plan Sponsors
by Christina Ha, Assistant Vice President
Lawsuits are commonplace nowadays, especially in the pharmacy industry. Recent headlines have been around the Purdue bankruptcy lawsuit to satisfy its creditors, and “Bad Drug” lawsuits where individuals sue pharmaceutical manufacturers for suffering injury due to undisclosed side effects, or because of manufacturing defects like contamination. The recent drug recalls of Zantac and valsartan are an example of the latter. But the types of lawsuits that typically affect third-party payors are the class-action lawsuits against pharmaceutical manufacturers for an action that has harmed them by increasing the cost they paid for pharmaceuticals.
What is a class action lawsuit and who is a third-party payor?
A class action lawsuit happens when several people suffer similar injuries as a result of the same product or same wrongful action. While it may not make sense for one person’s claims to support a lawsuit, when there is a group that bands together, the value of the lawsuit can start to add up. Third-party payors are self-insured employers, PBMs, Health Plans, traditional insurance companies, union benefit funds, and other forms of ERISA plans. Unlike the “Bad Drug” lawsuits, these types of lawsuits brought on by third-party payors are not claiming that drugs are unsafe, but allege that pharma manufacturers violated antitrust and consumer protection laws. In other words, they are increasing a payor’s cost by actions such as stifling generic competition from entering the market, fixing prices of drugs to keep them inflated, or promoting more utilization through off-label or misleading advertisement. Often the manufacturers agree to settle these lawsuits, for millions, sometimes hundreds of millions of dollars.
The Role of Generic Competition
When a new brand drug enters the market, the innovator manufacturer files for a patent that is good for 20 years, but typically only 7-12 years remain once it has reached the market. This allows the manufacturer a set amount of time to profit from the sale of the drug, since research and development costs to bring a new drug to market are staggering. Estimated costs range from $895M-$2 B. After this patent has expired, generic manufacturers can enter the market, which brings down the cost of both the brand and the generic drugs. The innovator manufacturer, not wanting to lose profits, can employ certain tactics such as “pay for delay” where the patent holders pay generic manufacturers to delay entry of the generic drugs to the market. The Federal Trade Commission (FTC) estimates that “pay for delay” tactics cost the taxpayers, insurance companies, and consumers ~$3.5B a year. Some of the most recent class action lawsuits that you may have heard of are for Thalomid/Revlimid (settled for $34M) and Provigil/Nuvigil (settled for $66M) where the defendants allegedly employed this tactic.
How do I know if I can take part in settlements from any current Pharmacy Class Action Lawsuits? What action is needed on my part?
Your PBM may proactively notify you of the lawsuit and provide the information needed to file a claim. Alternately, you may get a notice directly from the law firm handling the case who will notify you of your rights and actions needed in order to file a claim. In all cases, proof that you have paid for the drug(s) in question during a particular time frame is a requirement to participate in the settlement. The amount that employer groups will be paid depends on the settlement amount, the number of third-party payors that decide to participate, and how much was paid for the drugs in question. Some PBM’s will even file claims on their client’s behalf, while others will only provide a claim file and leave the rest of the work up to the client’s legal team.
Lockton partners with firms that can help navigate through the claim filing and settlement process. Your Lockton team can connect you with someone who can help with current and future class action lawsuits.

Christina Ha
Assistant Vice President
Christina joined LDB in 2015. As a senior account executive, Christina’s specialties include pharmacy benefit procurement, clients’ pharmacy utilization and program analysis, and performing contract reviews. She provides year-end audits of clients’ financial guarantees to ensure they were met or helps to get clients reimbursed if the PBM falls short. Christina works with cost forecasting, data analysis, plan design, specialty drug costs, and emerging pharmacy trends. She prepares and presents pharmacy deliverables to clients, including RFP results, ad hoc analysis, year-end audit reconciliations, and contract review recommendations. Additionally,
Christina assists with implementation.
Christina also works on the Specialty Pipeline Cost Predictor in Infolock®. She helps monitor the pipeline and is part of the workgroup that helps keep the Specialty Pipeline Cost Predictor up to date and current.